
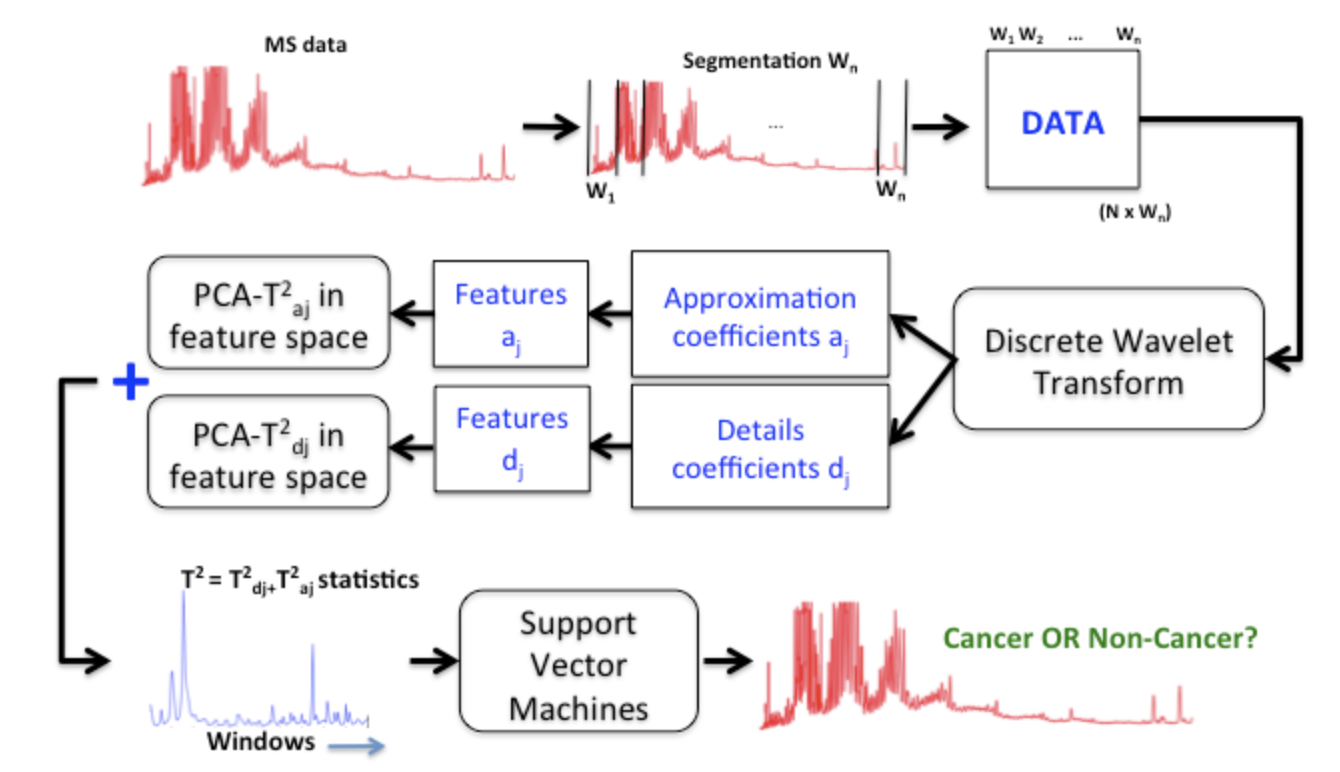
Overview
Proteomic patterns can help the diagnosis of the underlying pathological state of an organ such as the ovary, the lung, and the breast, to name few. An accurate classification of mass spectrometry is a crucial point to establish a reliable diagnosis and decision process regarding a type of cancer.
A statistical methodology for classifying mass spectrometry data is proposed. An overview of wavelets, principal component analysis-T2 statistic, and support vector machines is given. The study is performed on low-mass SELDI spectra derived from patients with breast cancer and from normal controls. There are 156 samples where control (normal) patients contribute with 57 samples and 99 samples are cancer. A hyperparameter optimization is conducted to select a support vector machine classification model based on grid search. The performance was evaluated with a k-fold cross validation technique and Monte-Carlo simulation with 100 replications. The average accuracy is 100% with standard error equals to 0. The averages of the sensitivity and specificity are both equal to 100%, as well as the area under the curve. The excellent performance of the proposed method is mainly due to the modeling, the new PCA-T2 statistic combining details and approximation coefficients, and the feature extraction procedure proposed (Cohen, Messaoudi, and Badir 2018).
References
Cohen, Achraf, Chaimaa Messaoudi, and Hassan Badir. 2018. “A New Wavelet-Based Approach for Mass Spectrometry Data Classification.” New Frontiers of Biostatistics and Bioinformatics, 175–89.